Molekülorbitaltheorie

Hans-Peter Ahlsen
Chemie,
Physikalische Chemie, MO-Methode, molecular orbital method, ein
quantenmechanisches Näherungsverfahren zur Berechnung der Elektronenstruktur
und der Energie von Molekülen, das im wesentlichen von Hund, Mulliken,
Lennard-Jones und Hückel (1927-29) entwickelt wurde. Dabei betrachtet man
analog dem Zentralfeldmodell der Atome ein Elektron, das sich in einem
effektiven Potential der Kerne und der übrigen Elektronen bewegt. Die daraus
resultierenden Einelektronenzustände des Moleküls bezeichnet man als
Molekülorbitale (Abk. MO) mit der Wellenfunktion F. Sie erstrecken sich
über das gesamte Molekül und sind durch eine Orbitalenergie gekennzeichnet. Die
Besetzung der energetisch niedrigsten Molekülorbitale unter Beachtung des
Pauli-Prinzips führt zur Elektronenkonfiguration der Moleküle im Grundzustand.
Die mathematische Darstellung von F als Linearkombination von
Atomorbitalen mit Wellenfunktionen yi stellt einen
geeigneten Näherungsansatz dar (MO-LCAO-Ansatz, Abk. von molecular orbitals -
linear combination of atomic orbitals): ![]() . Beim
Variationsverfahren werden die Koeffizienten ci
so gewählt, dass die Energie des Gesamtsystems einen Minimalwert annimmt.
. Beim
Variationsverfahren werden die Koeffizienten ci
so gewählt, dass die Energie des Gesamtsystems einen Minimalwert annimmt.
Das Prinzip der Molekülorbitalmethode lässt sich am einfachsten
für homonukleare zweiatomige Moleküle, z.B. für das ![]() -Molekül,
demonstrieren. Die Kombination der 1s-Orbitale der beiden Wasserstoffatome A
und B führt zu zwei Molekülorbitalen
-Molekül,
demonstrieren. Die Kombination der 1s-Orbitale der beiden Wasserstoffatome A
und B führt zu zwei Molekülorbitalen ![]() . Die
entsprechenden Orbitalenergien E+ und E- weisen gegenüber
der Energie der Atomorbitale eine niedrigere bzw. höhere Energie auf. Daher
bezeichnet man den Zustand F+ als bindendes und F- als lockerndes (antibindendes) Molekülorbital. Im
Grundzustand des Wasserstoffmoleküls besetzen beide Elektronen das bindende
Molekülorbital. Damit kann die chemische Bindung im Wasserstoffmolekül erklärt
werden.
. Die
entsprechenden Orbitalenergien E+ und E- weisen gegenüber
der Energie der Atomorbitale eine niedrigere bzw. höhere Energie auf. Daher
bezeichnet man den Zustand F+ als bindendes und F- als lockerndes (antibindendes) Molekülorbital. Im
Grundzustand des Wasserstoffmoleküls besetzen beide Elektronen das bindende
Molekülorbital. Damit kann die chemische Bindung im Wasserstoffmolekül erklärt
werden.
Analog zu den 1s-Orbitalen können auch andere Atomorbitale zu Molekülorbitalen kombinieren. Auf Grund der durch die Winkelteile bestimmten Symmetrie und Richtungsabhängigkeit der Atomorbitale unterscheidet man verschiedene Überlappungsmöglichkeiten. Die Kombination von rotationssymmetrisch zur Bindungsachse angeordneten Orbitalen liefert s-Molekülorbitale. Sie sind durch ein Überlappungsgebiet in der Bindungsachse gekennzeichnet. Die Wechselwirkung von Atomorbitalen, die eine Antisymmetrieebene in der Bindungsachse aufweisen, führt zu p-Molekülorbitalen. Sie bilden zwei Überlappungsgebiete ausserhalb der Bindungsachse, die durch eine Knotenebene getrennt sind. Treten vier Überlappungsgebiete ausserhalb der Bindungsachse auf, die durch zwei senkrecht zueinander stehende Knotenebenen getrennt sind, so liegt ein d-Molekülorbital vor. Die so gebildeten Bindungen werden als s, p- bzw. d-Bindung bezeichnet. Die jeweils lockernden Molekülorbitale werden zusätzlich durch einen Stern markiert, z.B. s*, p*, d*. Sie weisen neben den Knotenflächen der beteiligten Atomorbitale noch eine zusätzliche Knotenfläche zwischen den Atomen senkrecht zur Kernverbindungsachse auf.
Ausgehend von den Überlappungsmöglichkeiten der Atomorbitale kann die Molekülorbitalbehandlung auf homonukleare zweiatomige Moleküle von Elementen der 2. Periode (Li2, B2, C2, N2, O2, F2) ausgedehnt werden. Der MO-LCAO-Ansatz kann auch auf kompliziertere Moleküle angewendet werden, wobei der Rechenaufwand mit steigender Anzahl von Atomorbitalen stark zunimmt.
Mit einer Kombination aus Elektronenstrahl- und Röntgenbeugung konnten J.-M. Zuo, J.C.H. Spence und Kollegen die Molekülorbitale in Kupfer(I)oxid sichtbar machen (siehe Abb.) und nachweisen, dass dabei neben ionischen Bindungen auch kovalente Bindungen (chemische Bindung) sowie - überraschenderweise - Bindungen zwischen den Metallatomen auftreten.
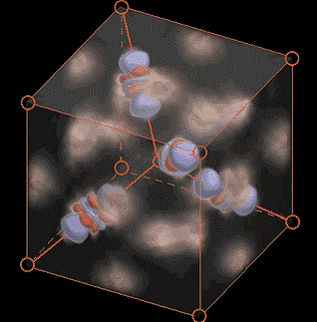
Molekülorbitaltheorie: In Messungen an Cuprit (CU2O) gefundene kompliziert geformte Orbitale, die aus der Hybridisierung von einem s- und zwei dz-Orbitalen hervorgegangen sind. Ihr Aussehen entspricht genau den theoretischen Berechnungen: eine Hantel mit einem Ring und zwei weiteren Ringen (J.M. Zuo, M. Kim, M. O\'Keeffe und J.C.H. Spence: Nature 401, 49 (1999)).
Techniklexikon.net
Das freie Technik-Lexikon. Fundierte Informationen zu allen Fachgebieten der Ingenieurwissenschaften, für Wissenschaftler, Studenten, Praktiker & alle Interessierten. Professionell dargeboten und kostenlos zugängig.
Techniklexikon
Modernes Studium der Physik sollte allen zugängig gemacht werden.