Dichtefunktionaltheorie

Irene Kramer-Schwenk
Atom- und Molekülphysik
bottom:.0001pt\'>1 Einführung
bottom:.0001pt\'>Die Dichtefunktionaltheorie (DFT) [1,2] kann als die exakte Darstellung des quantenmechanischen Vielteilchenproblems durch die Einteilchendichte n(r) bezeichnet werden. Ihre Wurzeln reichen mit der Thomas-Fermi(TF)-Theorie (Thomas-Fermi-Näherung) als der einfachsten Theorie quantenmechanischer Vielteilchensysteme bis in die Anfangszeit der Quantenmechanik zurück. Die TF-Theorie ermöglicht zwar mit relativ wenig Aufwand die Gewinnung formal exakter Ergebnisse, der Vergleich mit den Eigenschaften realer elektronischer Systeme offenbart aber eine Reihe schwerwiegender Defekte der Theorie. So hat die durch direkte Integration der TF-Gleichung erhaltene Dichte n(r) das falsche asymptotische Verhalten für kleine und grosse r, die für die atomaren Elektronendichten charakteristische Schalenstruktur wird nicht erhalten, und vor allem kann die Existenz kondensierter Materie nicht deduziert werden. Erweiterungen der TF-Theorie durch Dirac mit der Berücksichtigung des Austauschs, durch Fermi und Amaldi mit der Korrektur der Selbstwechselwirkung in einer gemittelten Form, durch Weizsäcker mit der Verbesserung der kinetischen Energie über eine Gradientenentwicklung, sowie zahlreiche, meist phänomenologische Modifikationen zeigten in quantitativer Hinsicht nur bescheidene Erfolge. Die Einsicht, dass viele Defekte der TF-Theorie in der schlechten Darstellung der kinetischen Energie begründet sind, führte 1951 zuerst Slater zur Formulierung eines Ansatzes, in dem die kinetische Energie entsprechend der Hartree-Fock(HF)-Theorie (Hartree-Fock-Näherung) mit Hilfe von Wellenfunktionen berechnet, der Austausch aber entsprechend der TF-Theorie über alle besetzten Zustände gemittelt wurde. Die Verallgemeinerung dieses Ansatzes zur sog. Xa-Näherung lieferte mit der Hartree-Fock-Slater(HFS)-Theorie (Hartree-Fock-Slater-Näherung) den Rahmen für eine ganze Klasse von Methoden zur Berechnung der elektronischen Struktur von Molekülen und Festkörpern [3]. Deren praktische Erfolge in den siebziger und frühen achtziger Jahren wurden in Anbetracht der relativen Einfachheit dieses theoretischen Schemas allgemein als überraschend, wenn nicht gar als reiner Zufall angesehen. Dass die guten Ergebnisse der HFS-Theorie kein Zufall sind und auch die TF-Theorie keine fiktive, für praktische Zwecke irrelevante Modelltheorie darstellt, zeigt die Integration beider Ansätze in die DFT; die DFT kann insofern als die exakte, quantenmechanische Erweiterung der TF-Theorie angesehen werden.
bottom:.0001pt\'>Die zentrale Aussage
der DFT, dass die Grundzustandseigenschaften eines quantenmechanischen
Vielteilchensystems allein durch seine Einteilchendichte n(r) eindeutig und vollständig bestimmt sind, ist durchaus
bemerkenswert und keineswegs selbstverständlich, insbesondere vor dem
Hintergrund "konventioneller" Darstellungen des quantenmechanischen
Vielteilchenproblems. Darunter sind alle Darstellungen zu verstehen, deren
zentrale Grösse die Vielteilchenwellenfunktion Y ist. In diesem Rahmen
können die Vielteilcheneffekte mit Hilfe der Paardichte (Korrelation)
beschrieben werden, die symbolisch in der Form ![]() darstellbar ist, wobei
darstellbar ist, wobei ![]() die Paarkorrelationsfunktion bezeichnet. In
HF-Näherung ist
die Paarkorrelationsfunktion bezeichnet. In
HF-Näherung ist ![]() entsprechend dem Modell unabhängiger Teilchen,
während etwa die Entwicklung von Y nach Slater-Determinanten dazu führt,
dass die Paardichte nicht mehr faktorisierbar ist. Insofern kann man sagen, dass
in der konventionellen Darstellung des Vielteilchenproblems alle
Vielteilcheneffekte in der Paardichte enthalten sind. Es ist verständlich, dass
vor diesem Hintergrund jede Theorie, die mit der Einteilchendichte auszukommen
behauptet, als nicht exakt erscheinen muss; dass dies nicht der Fall ist, gehört
zu den grundlegenden Ergebnissen der DFT.
entsprechend dem Modell unabhängiger Teilchen,
während etwa die Entwicklung von Y nach Slater-Determinanten dazu führt,
dass die Paardichte nicht mehr faktorisierbar ist. Insofern kann man sagen, dass
in der konventionellen Darstellung des Vielteilchenproblems alle
Vielteilcheneffekte in der Paardichte enthalten sind. Es ist verständlich, dass
vor diesem Hintergrund jede Theorie, die mit der Einteilchendichte auszukommen
behauptet, als nicht exakt erscheinen muss; dass dies nicht der Fall ist, gehört
zu den grundlegenden Ergebnissen der DFT.
bottom:.0001pt\'>
bottom:.0001pt\'>2 Der theoretische Rahmen
bottom:.0001pt\'>2.1 Die Hohenberg-Kohn-Theoreme
bottom:.0001pt\'>Als Geburtsjahr der DFT kann das Jahr 1964 gelten, in dem mit einer Arbeit von Hohenberg und Kohn [4] die theoretische Basis und der konzeptionelle Rahmen dieser Theorie in Form einer Reihe von Sätzen begründet wurde, die heute den Namen Hohenberg-Kohn(HK)-Theoreme tragen. Um deren Bedeutung und Tragweite verständlich zu machen, sei daran erinnert, dass in der konventionellen Behandlung des N-Elektronenproblems das von der Kernanordnung erzeugte "externe" Potential v ext(r) zusammen mit der Elektronenzahl N den Hamilton-Operator und damit im Prinzip alle Eigenschaften des Grundzustands eindeutig festlegt. Das erste HK-Theorem besagt nun in seiner ursprünglichen Formulierung, dass im Fall eines nichtentarteten Grundzustands statt N und v ext(r) auch die Einteilchendichte n(r) dies leistet:
bottom:.0001pt\'>(HK1) Das externe Potential v ext(r) eines N-Elektronensystems ist bis auf eine physikalisch bedeutungslose Konstante eindeutig durch die Einteilchendichte n(r) bestimmt. Die Energie E0 des Grundzustands ist ein eindeutiges Funktional dieser Dichte, d.h. E0 = E[n].
bottom:.0001pt\'>Da die
Einteilchendichte n(r) über die
Normierungsbedingung ![]() die Zahl der Elektronen festlegt, bestimmt sie
im Prinzip auch die Wellenfunktion des Grundzustands. Ferner ist das externe
Potential v ext(r) nicht auf Coulomb-Potentiale beschränkt; es muss
lediglich lokal sein und kann daher auch entsprechende von aussen angelegte
elektrische Felder einschliessen. Der Beweis dieses Theorems ist erstaunlich
einfach und beruht auf reductio ad absurdum: es werden zwei wesentlich,
d.h. um mehr als eine Konstante verschiedene, externe Potentiale angenommen,
die zu derselben Grundzustandsdichte gehören sollen; anschliessend wird gezeigt,
dass diese Annahme auf einen Widerspruch führt. Es besteht daher ein umkehrbar
eindeutiger Zusammenhang zwischen n(r)
und v ext(r). Darüber hinaus gilt für die Einelektronendichte ein
Variationsprinzip, in dem die Grundzustandsdichte das Funktional E[n]
der Gesamtenergie stationär macht:
die Zahl der Elektronen festlegt, bestimmt sie
im Prinzip auch die Wellenfunktion des Grundzustands. Ferner ist das externe
Potential v ext(r) nicht auf Coulomb-Potentiale beschränkt; es muss
lediglich lokal sein und kann daher auch entsprechende von aussen angelegte
elektrische Felder einschliessen. Der Beweis dieses Theorems ist erstaunlich
einfach und beruht auf reductio ad absurdum: es werden zwei wesentlich,
d.h. um mehr als eine Konstante verschiedene, externe Potentiale angenommen,
die zu derselben Grundzustandsdichte gehören sollen; anschliessend wird gezeigt,
dass diese Annahme auf einen Widerspruch führt. Es besteht daher ein umkehrbar
eindeutiger Zusammenhang zwischen n(r)
und v ext(r). Darüber hinaus gilt für die Einelektronendichte ein
Variationsprinzip, in dem die Grundzustandsdichte das Funktional E[n]
der Gesamtenergie stationär macht:
bottom:.0001pt\'>(HK2) Gegeben sei eine
Elektronendichte n ¢ (r), die die
Bedingungen n ¢ (r) ³ 0
(für alle r) und ![]() erfülle. Dann ist die damit berechnete Energie
immer grösser als die exakte Grundzustandsenergie E0, d.h. E[n ¢ ] ³ E0, und das
Gleichheitszeichen gilt genau dann, wenn n ¢ (r) gleich der exakten Einteilchendichte n(r) des Grundzustands ist.
erfülle. Dann ist die damit berechnete Energie
immer grösser als die exakte Grundzustandsenergie E0, d.h. E[n ¢ ] ³ E0, und das
Gleichheitszeichen gilt genau dann, wenn n ¢ (r) gleich der exakten Einteilchendichte n(r) des Grundzustands ist.
bottom:.0001pt\'>Ist ausserdem E[n] differenzierbar, lässt sich unter der Nebenbedingung fester Teilchenzahl N die Euler-Lagrange-Gleichung
bottom:.0001pt\'>![]() (1)
(1)
bottom:.0001pt\'>herleiten, wobei der Lagrange-Parameter m als chemisches Potential interpretiert werden kann. Die Existenz des Funktionals E[n] ist dabei aufgrund von (HK1) zwar gesichert, über dessen konkrete Form gibt das Theorem aber keine Auskunft.
bottom:.0001pt\'>Einen ersten Schritt in diese Richtung stellt das dritte HK-Theorem dar:
bottom:.0001pt\'>(HK3) Die Gesamtenergie eines N-Elektronensystems in einem lokalen, externen Potential v ext(r) kann dargestellt werden in der Form
bottom:.0001pt\'>![]() (2)
(2)
bottom:.0001pt\'>mit einem von v ext(r) unabhängigen und in diesem Sinn universellen Funktional FHK[n], das sich aus der kinetischen Energie T[n] der Elektronen und der Elektron-Elektron-Wechselwirkung Vee[n] zusammensetzt.
bottom:.0001pt\'>In Bezug auf seine n-Abhängigkeit hat demnach FHK[n] in allen Systemen dieselbe Form, so dass das N-Elektronenproblem in einen allgemeinen, systemunabhängigen und in einen systemspezifischen Teil zerlegt werden kann. Der allgemeine Teil des Problems hat die Frage zu klären, in welcher Form FHK von n(r) abhängt, während der systemspezifische Teil ausschliesslich durch die spezielle Form von n(r) bestimmt ist: beim Übergang von einem elektronischen System zu einem anderen ändert sich folglich nur n(r), nicht dagegen die funktionale Form von FHK. Diese Zerlegung der Gesamtenergie ist von fundamentaler Bedeutung, da sie im Prinzip die Behandlung des N-Elektronenproblems unabhängig von Annahmen über spezielle Systeme ermöglicht.
bottom:.0001pt\'>
bottom:.0001pt\'>2.2 Die Kohn-Sham-Darstellung
bottom:.0001pt\'>Über die konkrete Form von FHK als Funktional von n(r) wird in den Hohenberg-Kohn-Theoremen keine Aussage gemacht. Die Bestimmung dieses Funktionals ist eine der zentralen Aufgaben der DFT, weil dies nicht nur die notwendige Voraussetzung für praktische Anwendungen schafft, sondern gleichbedeutend mit der Lösung des Vielteilchenproblems ist. Die grösste Schwierigkeit stellt dabei die kinetische Energie T[n] dar, weil sie einen grossen Beitrag zur Gesamtenergie liefert und deswegen sehr genau berechnet werden muss. Da eine direkte Berechnung, wie sie im Rahmen der TF-Theorie versucht wird, zu schlechte Ergebnisse liefert, um für praktisch relevante Anwendungen konkurrenzfähig zu sein, schlugen Kohn und Sham [5] einen Lösungsansatz vor, der heute die Basis der meisten numerischen Rechnungen im Rahmen der DFT bildet. Die Grundidee dieser Kohn-Sham(KS)-Darstellung besteht darin, die kinetische Energie T0[n] des wechselwirkungsfreien N-Elektronensystems, die den bei weitem grössten Beitrag zur kinetischen Energie liefert, in exakter Form darzustellen und die verbleibende kleine Korrektur Tc[n] getrennt zu behandeln. Die Anwendung des Variationsprinzips führt dann auf eine Euler-Lagrange-Gleichung der Form
bottom:.0001pt\'> ![]() (3)
(3)
bottom:.0001pt\'>wobei das lokale, effektive Potential
bottom:.0001pt\'> ![]() (4)
(4)
bottom:.0001pt\'>als einzigen, nicht exakt bestimmbaren Beitrag das Austauschkorrelationspotential v xc[n(r)] (Austauschkorrelationsenergie) enthält. Diese Darstellung des quantenmechanischen Vielteilchenproblems hat die Struktur eines Problems unabhängiger Teilchen, ist aber unverändert exakt. Auch damit ist allerdings die direkte Lösung von Gleichung (3) nicht möglich, weil die explizite Form von T0[n] als Funktional von n(r) bisher nicht bekannt ist. Geht man aber zur Orbitaldarstellung der kinetischen Energie T0[n] über und führt die Variation nach den Orbitalen statt nach der Dichte durch, erhält man die kanonische Form der Kohn-Sham(KS)-Gleichungen in Orbitaldarstellung:
bottom:.0001pt\'>![]()
bottom:.0001pt\'>Diese Gleichungen sind nichtlinear und müssen iterativ gelöst werden, wobei die Selbstkonsistenz zwischen der Einteilchendichte n(r) und v eff(r), Gleichung (4), zu erreichen ist. Sie haben unverändert die Struktur von Einteilchengleichungen, sind aber nichtsdestoweniger eine exakte Darstellung des N-Elektronenproblems, wobei alle Vielteilcheneffekte in v xc[n(r)] enthalten sind. Als der entscheidende Vorteil der KS-Darstellung im Vergleich zur direkten Berechnung von T[n] erweist sich dabei, dass der Übergang zur Orbitaldarstellung die zwar indirekte, aber exakte Behandlung des grössten Teils T0 der kinetischen Energie ermöglicht. Dadurch werden nämlich die wesentlichen Defekte der TF-Theorie, wie das falsche asymptotische Verhalten von n(r), das Fehlen der Schalenstruktur bei Atomen sowie der chemischen Bindung in Molekülen und Festkörpern, beseitigt. Der Preis, den man für diese Verbesserung zu zahlen hat, ist allerdings, dass nun N Gleichungen für yi(r) zu lösen sind statt einer einzigen für n(r). Dennoch bleibt der Aufwand für die Lösung der KS-Gleichungen vergleichbar mit dem des quasiklassischen Hartree-Problems.
bottom:.0001pt\'>
bottom:.0001pt\'>2.3 Dichtefunktionaltheorie im Vergleich zur Wellenfunktionstheorie
bottom:.0001pt\'>Zusammenfassend lässt sich feststellen, dass die DFT eine Darstellung des quantenmechanischen N-Elektronenproblems liefert, die in gewisser Weise reziprok zu den konventionellen Ansätzen der Wellenfunktionstheorie (WFT) ist, in symbolischer Darstellung:
bottom:.0001pt\'>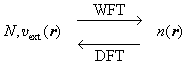 .
.
bottom:.0001pt\'>Zentrale Grösse der DFT
ist die Einteilchendichte n(r) des
Grundzustands, die im Gegensatz zur abstrakten Vielteilchenwellenfunktion Y
eine Observable und als Funktion im dreidimensionalen Ortsraum leicht
interpretierbar ist. Der Hamilton-Operator des N-Elektronensystems wird
in der KS-Darstellung der DFT in ein lokales Funktional der Einteilchendichte
transformiert und hat dadurch die Struktur eines Einteilchenoperators. Die
HF-Gleichungen enthalten dagegen mit dem HF-Austauschpotential einen
wesentlichen nichtlokalen Term und sind daher nicht als Spezialfall der
KS-Gleichungen rekonstruierbar. Als System von Einteilchen-Gleichungen sind die
KS-Gleichungen numerisch relativ leicht zu lösen, bilden aber unverändert eine
exakte Darstellung des N-Elektronenproblems. Dass dies kein Widerspruch
ist, hat seinen Grund darin, dass empirisch signifikant nur der Erwartungswert ![]() ist, nicht dagegen y oder H allein.
Es spielt daher keine Rolle, ob die Vielteilcheneffekte wie in der WFT über y
berücksichtigt werden, oder wie in der DFT über den Hamilton-Operator H,
der in Form des Austauschkorrelationsfunktionals sämtliche Vielteilcheneffekte
enthält. Neben dessen Berechnung, die eine der zentralen Aufgaben darstellt,
umfasst die aktuelle Forschung innerhalb der DFT die Untersuchung von
Grundlagenproblemen mit dem Ziel einer Verbesserung der theoretischen Basis,
Erweiterungen der Theorie etwa auf die Beschreibung angeregter Zustände bis hin
zu der von nichtelektronischen Systemen, sowie die praktischen Anwendungen in
Form der Berechnung von n(r) für reale
Systeme wie Atome, Moleküle und Festkörper.
ist, nicht dagegen y oder H allein.
Es spielt daher keine Rolle, ob die Vielteilcheneffekte wie in der WFT über y
berücksichtigt werden, oder wie in der DFT über den Hamilton-Operator H,
der in Form des Austauschkorrelationsfunktionals sämtliche Vielteilcheneffekte
enthält. Neben dessen Berechnung, die eine der zentralen Aufgaben darstellt,
umfasst die aktuelle Forschung innerhalb der DFT die Untersuchung von
Grundlagenproblemen mit dem Ziel einer Verbesserung der theoretischen Basis,
Erweiterungen der Theorie etwa auf die Beschreibung angeregter Zustände bis hin
zu der von nichtelektronischen Systemen, sowie die praktischen Anwendungen in
Form der Berechnung von n(r) für reale
Systeme wie Atome, Moleküle und Festkörper.
bottom:.0001pt\'>
bottom:.0001pt\'>2.4 Theorieerweiterungen
bottom:.0001pt\'>In ihrer ursprünglichen Formulierung bezieht sich die DFT auf den Grundzustand eines N-Elektronensystems in einem zeitunabhängigen, lokalen, externen Potential ohne Berücksichtigung magnetischer und relativistischer Effekte. Mittlerweile existieren jedoch eine ganze Reihe von Erweiterungen dieses "Theoriekerns", wie eine zeitabhängige und eine relativistische DFT, eine DFT der Supraleitung sowie Formulierungen für mehrkomponentige und nichtelektronische Systeme.
bottom:.0001pt\'>Die für praktische
Anwendungen besonders wichtige Erweiterung der KS-Gleichungen auf Systeme mit
offenen Schalen ist im Rahmen einer nichtrelativistischen Beschreibung leicht
möglich. Enthält der Hamilton-Operator keine spinabhängigen Wechselwirkungen
und exterene Magnetfelder, kann man ganz analog zum spinfreien Fall ein System
von spinabhängigen ("spin-polarisierten") KS-Gleichungen getrennt für
![]() und
und ![]() herleiten. Die daraus resultierende
Erweiterung wird als Spindichtefunktionaltheorie (SDFT) bezeichnet. Eine andere
wichtige Erweiterung der DFT betrifft die elektronisch angeregten Zustände. Für
die allgemeine Lösung dieses Problems existiert zwar eine Reihe von Ansätzen
[1, 2], insgesamt muss man jedoch sagen, dass eine formal befriedigende und
gleichzeitig praktisch nutzbare Beschreibung angeregter Zustände bisher nicht
existiert, obwohl im pragmatischen Sinn die DFT in der Regel für die Berechnung
mindestens der niedrigsten angeregten Zustände funktioniert.
herleiten. Die daraus resultierende
Erweiterung wird als Spindichtefunktionaltheorie (SDFT) bezeichnet. Eine andere
wichtige Erweiterung der DFT betrifft die elektronisch angeregten Zustände. Für
die allgemeine Lösung dieses Problems existiert zwar eine Reihe von Ansätzen
[1, 2], insgesamt muss man jedoch sagen, dass eine formal befriedigende und
gleichzeitig praktisch nutzbare Beschreibung angeregter Zustände bisher nicht
existiert, obwohl im pragmatischen Sinn die DFT in der Regel für die Berechnung
mindestens der niedrigsten angeregten Zustände funktioniert.
bottom:.0001pt\'>
bottom:.0001pt\'>2.5 Konstruktion von Funktionalen
bottom:.0001pt\'>Die Austauschkorrelationsenergie Exc ist in der KS-Darstellung definiert als Differenz zwischen der exakten Grundzustandsenergie und denjenigen Anteilen, die exakt berechenbar sind. Eine exakte analytische Darstellung auch des Austauschkorrelationsfunktionals Exc[n] kann dagegen nicht erwartet werden, denn das würde ja bedeuten, dass das quantenmechanische N-Elektronenproblem analytisch lösbar wäre. Da eine solche Lösung nicht einmal für das freie oder homogene Elektronengas als dem einfachsten elektronischen System existiert, muss man zwangsläufig auf Näherungen zurückgreifen. Die einfachste ist die sog. Lokale-Dichte- bzw. die Lokale-Spin-Dichte (LSD)-Näherung, in deren Rahmen die Austauschkorrelationsenergie eines beliebigen, inhomogenen Systems lokal, d.h. in einem infinitesimal kleinen Volumen d3r wie die eines homogenen Systems angenommen wird. Trotz ihrer zahlreichen Erfolge [3, 6] enthält die LSD-Näherung eine Reihe charakteristischer Schwächen, die zu einem grossen Teil auf die unzureichende Darstellung des Austauschbeitrages zu Exc[n] zurückzuführen sind. Die meisten dieser Defekte lassen sich jedoch durch Austauschkorrelationfunktionale beseitigen, die Gradientenkorrekturen erster Ordnung in n(r) einschliessen (Gradienten-Näherung). Damit konnten in den letzten Jahren grosse Fortschritte, insbesondere bei der Berechnung von Bindungsenergien und damit der thermochemischen Charakterisierung von Molekülen erzielt werden [7]. Ein grosses Problem stellen dagegen nach wie vor die stark korrelierten Systeme dar, was insofern verständlich ist, als die gradientenkorrigierten Funktionale von einer Verbesserung der LSD-Näherung ausgehen. Fortschritte in diesem Bereich dürften deswegen eher von einem alternativen Ansatz zu erwarten sein, bei dem Austauschkorrelationsfunktionale ausgehend von der Dichte-Matrix-Darstellung der WFT konstruiert werden.
bottom:.0001pt\'>
bottom:.0001pt\'>3 Anwendungen
bottom:.0001pt\'>Der ursprüngliche Anwendungsbereich numerischer DFT-Methoden, wenn man von Anwendungen der TF-Theorie absieht, ist die Festkörperphysik. Die ersten Bandstrukturrechnungen mit dem Slaterschen Austauschpotential (Hartree-Fock-Slater-Näherung) datieren aus den fünfziger Jahren, und heute erfolgen schätzungsweise mehr als 90 % aller Berechnungen der elektronischen Struktur von Festkörpern, Oberflächen und dünnen Schichten im Rahmen der DFT. Während bis etwa Anfang der achtziger Jahre überwiegend die Lokale-Dichte-Näherung verwendet wurde, gehören heute Rechnungen mit gradientenkorrigierten Funktionalen zum Standard. Erste Rechnungen an Molekülen sind erst Ende der sechziger Jahre versucht worden, und bis Mitte der achtziger Jahre stellten die DF-Methoden eher eine Randerscheinung innerhalb der traditionellen Quantenchemie dar. Die bis dahin durchweg verwendeten LSD-Methoden lieferten zwar recht gute Ergebnisse für die elektronische Struktur und zahlreiche spektroskopische Daten, ergaben aber erheblich zu grosse Bindungsenergien und waren damit für die thermochemische Charakterisierung von Molekülen nicht geeignet. Dies änderte sich erst mit dem Einsatz gradientenkorrigierter oder noch allgemeinerer ("nichtlokaler") Austauschkorrelationsfunktionale; seither zählen die DF-Methoden auch in der Quantenchemie zu den am meisten verwendeten Verfahren. Aus diesen Gründen ist eine repräsentative Beschreibung der numerischen Anwendungen der DFT in der Atom-, Molekül- und Festkörperphysik mit wenigen Sätzen nicht mehr möglich. Im grossen und ganzen kann man jedoch sagen, dass die Berechnung messbarer Grössen wie Geometrien, Dipolmomente, Ionisierungspotentiale, Bindungsenergien, Schwingungsfrequenzen und Hyperfein-Parameter bereits in LSD-Näherung oftmals bessere Ergebnisse liefert als die HF-Näherung, während mit den gegenwärtig zur Verfügung stehenden nichtlokalen Funktionalen in den meisten Fällen eine mit den besten Vielteilchenrechnungen vergleichbare Güte erreicht werden kann. Die Vorteile der DF-Methoden werden besonders deutlich bei den Verbindungen der Übergangsmetalle, bei denen HF-Methoden häufig qualitativ falsche Resultate liefern und die Anwendung von Vielteilchenmethoden sehr aufwendig ist, sowie bei der Behandlung grosser Systeme wie biologischer Moleküle. Schliesslich sind die in letzter Zeit stark zunehmenden Untersuchungen zur Charakterisierung chemischer Reaktionen zu erwähnen, sowie die erfolgreiche Kombination von DF-Methoden mit der Moleküldynamik zur Bestimmung von Strukturen kleiner Cluster [6]. Bei allen diesen Erfolgen sollte jedoch nicht übersehen werden, dass die DFT noch weit davon entfernt ist, das Allheilmittel für die Bewältigung sämtlicher Probleme der Elektronentheorie von Atomen, Molekülen, Clustern und Festkörpern zu sein.
bottom:.0001pt\'>
bottom:.0001pt\'>4 Zusammenfassung
bottom:.0001pt\'>Die DFT liefert eine formal exakte Darstellung des quantenmechanischen Vielteilchenproblems, die in gewisser Weise komplementär zu den traditionellen Methoden der Wellenfunktionstheorie (WFT) ist. Die Entwicklungen der letzten 30 Jahre haben dabei gezeigt, dass in dieser Umformulierung des Vielteilchenproblems sowohl in praktischer als auch in konzeptioneller Hinsicht ein enormes Potential liegt. Denn die Existenz einer exakten Theorie, in der die Einteilchendichte die Schlüsselrolle spielt, die im Gegensatz zur Vielteilchenwellenfunktion observabel und leicht interpretierbar ist, liefert nicht nur eine ganze Familie effizienter Berechnungsverfahren, sondern vor allem einen begrifflichen Rahmen, der den empirischen Begriffssystemen der Physik und Chemie wesentlich angepasster ist als das abstraktere Begriffssystem der WFT. Die Popularität und Verbreitung der DFT bei der Untersuchung von Elektronenstrukturproblemen dürfte aus diesen Gründen auch in der Zukunft weiter zunehmen.
Literatur[1] R.G. Parr, W. Yang: Density-Functional Theory of Atoms and Molecules, Oxford University Press, Oxford 1989.
[2] R.M. Dreizler, E.K.U. Gross: Density Functional Theory; Springer, Heidelberg-New York 1990.
[3] J.P. Dahl, J. Avery (Eds.): Local Density Approximations in Quantum Chemistry and Solid State Physics, Plenum Press, New York-London, 1984.
[4] P. Hohenberg, W. Kohn, Phys. Rev. 136 (1964) B864-871.
[5] W. Kohn, L.J. Sham, Phys. Rev. 140 (1965) A1133-1138.
[6] R.O. Jones, O. Gunnarsson, Rev. Mod. Phys. 61(1989) 689-746.
[7] R.F. Nalewajski (Hrsg.), Density Functional Theory I; Top. Curr. Chem. 180, Springer 1996.
Techniklexikon.net
Das freie Technik-Lexikon. Fundierte Informationen zu allen Fachgebieten der Ingenieurwissenschaften, für Wissenschaftler, Studenten, Praktiker & alle Interessierten. Professionell dargeboten und kostenlos zugängig.
Techniklexikon
Modernes Studium der Physik sollte allen zugängig gemacht werden.